ChIP-seq pipeline steps and reports
Analysis pipeline steps
Reads trimming: Reads are trimmed using cutadapt (DOI: 14806/ej.17.1.200) (with the parameters –times 2 -q 20 -m 25). In this process, primers corresponding to the Tru-seq protocol are removed.
Quality control: Reads quality control is evaluated using FastQC (with the parameter –cassava). A report file, containing quality results for all of the samples is generated using multiQC.
Mapping to genomes: The quality trimmed reads are mapped to Mouse and Human genomes using Bowtie2 (doi: 10.1038/nmeth.1923.) (with the parameters –local for all analyses and, in addition, -X 2000 for paired end analyses). Refseq annotation is provided for the mapped genes.
4. Alignment filtering: Following the alignment, reads are filtered using samtools view with the parameters -F 4 to remove unmapped reads that are output by bowtie2, -q 39 and with -f 0x2 for paired end reads. The remaining unique reads are then indexed and sorted, using samtools index and samtools sort.
Generation of statistics on the alignment using flagstat.
Visualization in graphs: The reads are graphically visualized using ngsplot (with the parameters -G -R genebody -C -O samples -D refseq -L 50000).
7. Peak calling: Significant chip regions (peaks) are evaluated and compared to control samples if present using macs2 callpeak (with the parameters –bw 300 -B -f –SPMR -g -keep-dup auto -q 0.01 for all analyses, BAMPE –nomodel for paired end analyses, and BAM for single end analyses). The resulting peaks “*_peaks_filtered.broadPeak” are filtered to exclude peaks from the blacklist (https://github.com/Boyle-Lab/Blacklist/tree/master/lists).
Conversion to BigWig format: Files containing the predicted peaks coordinates in BedGraph format are converted to BigWig format using bedtools slop (with the parameters -g -b 0), bedClip stdin and bedGraphToBigWig (with default parameters).
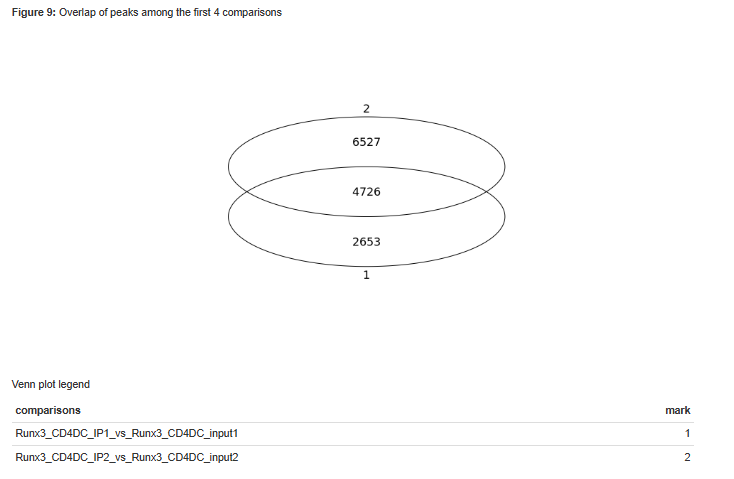
9. Peak annotation: The predicted peaks are collected from all samples using multiIntersectBed and then annotated according to the corresponding genome using Homer (with default parameters). Analysis of peaks distribution in genomic regions, and around TSS is done using ChipSeeker, together with a Venn diagram of overlap of peaks sets.

Pipeline report
Upon completion of the analysis, you will be sent an email with links to the results report.
The report includes several sections:
Sequencing and Mapping QC
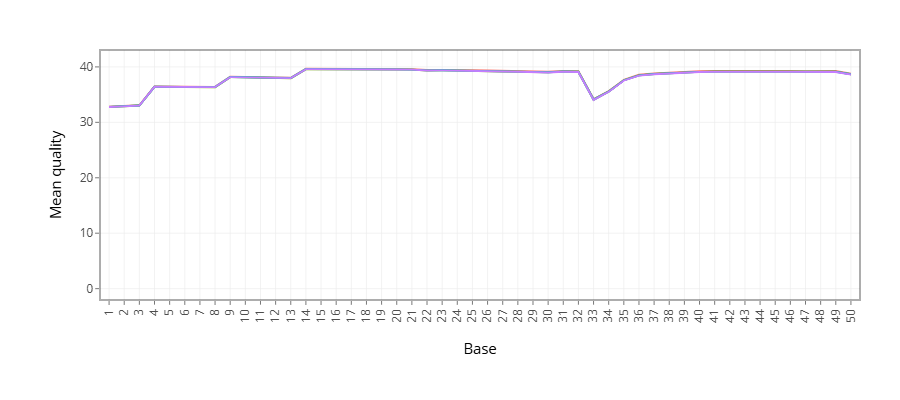
Figure 1 - Plots the average quality of each base across all reads. Qualities of 30 (predicted error rate 1:1000) and above are good.
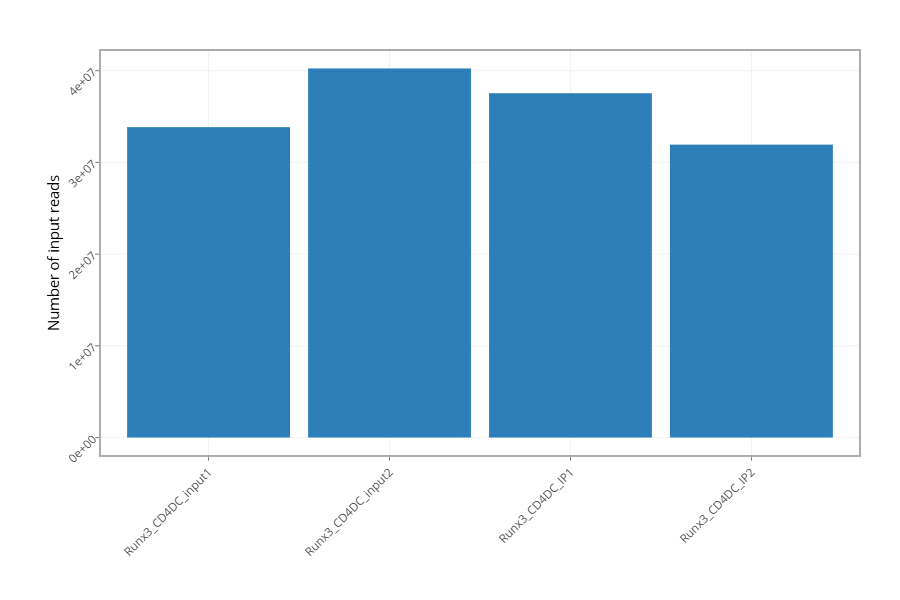
Figure 2 - Histogram showing the number of reads for each sample in the raw data.
Figure 3 - Histogram showing the percentage of reads discarded after trimming the adapters (after removing adapters, short, polyA/T and low quality reads are discarded by the pipeline). No figure will be presented if the percentage of reads discarded after trimming for all samples is lower than 1%.
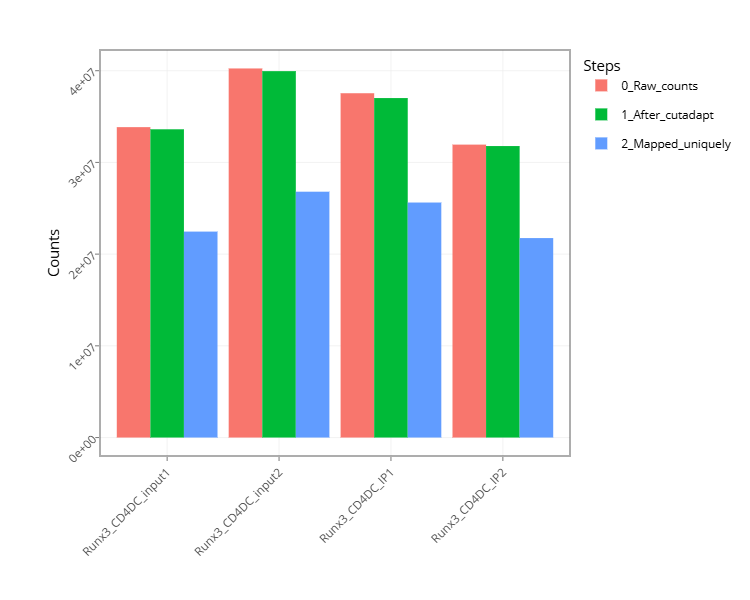
Figure 4 - Histogram with the number of reads for each sample in each step of the pipeline.
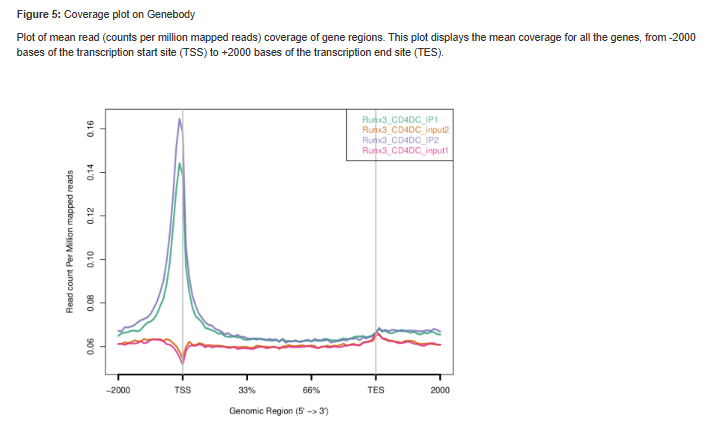
Figure 5 - Plots sequence coverage on and near gene regions.
- MACS peak calling
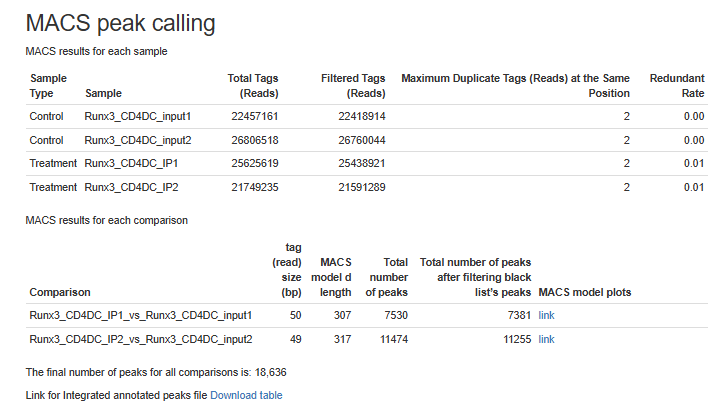
Figure 6 - MACS (peak calling) results table for each sample or for each comparison, if the pipeline was run with a control.
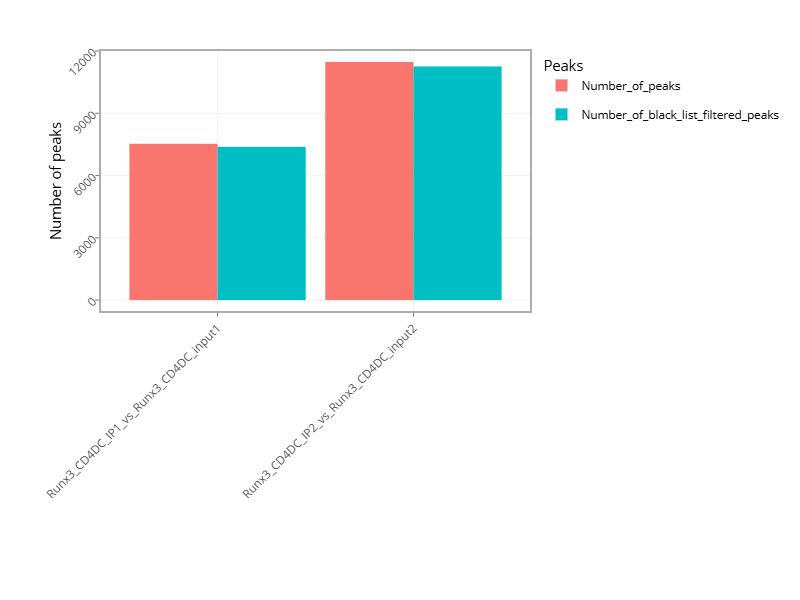
Figure 7 - Histogram with the number of peaks for all samples or for comparisons, if the pipeline was run with a control.
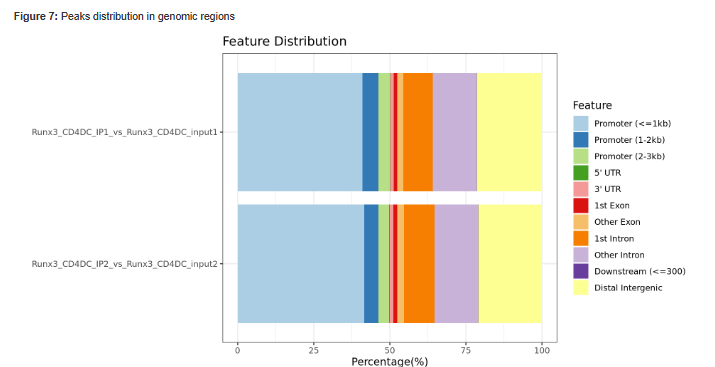
Figure 8 - Histogram showing Peaks distribution in genomic regions.
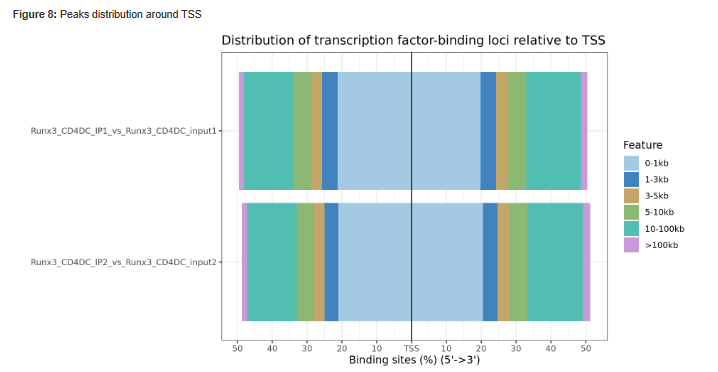
Figure 9 - Histogram showing Peaks distribution around TSS.
Figure 10 - Venn diagram of peak overlaps among the first four comparisons.
Bioinformatics Pipeline Methods - Description of pipeline methods.
Links to additional results - Links for downloading tables with raw, normalized counts, log normalized values (rld), and statistical data of contrasts.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Output folders
0_concatenating_fastq
1_cutadapt
2_fastqc
3_multiQC
4_mapping
5_filtered_alignment
6_peaks_prediction
7_peaks_annotation
8_graphs
9_BigWig
10_reports
Log directory
Annotation file
For Peak annotation, we use annotation files (gtf format) from “Ensembl” or “GENCODE”.